











. DOI: 10.1016/j.cell.2022.09.028")
Copy number variants (CNVs) are regions of the genome that are duplicated or deleted in some individuals, and are a common type of gene-disabling mutation. The human genome contains hundreds of thousands of CNVs, but typical genomic analysis approaches detect only the largest, and scientists aren’t sure what most of them do.
Now a team of researchers at the Broad Institute of MIT and Harvard, Brigham and Women’s Hospital, and Harvard Medical School has developed a computational method that detected 15 million CNVs in the U.K. Biobank—six times more than previous analyses of the same data. The researchers used their method to uncover hundreds of biological connections between these CNVs and dozens of human traits, revealing new links between specific genes and traits such as height, blood counts, and biological markers of health.
The findings, published today in Cell, are from the most thorough analysis of connections between CNVs and traits to date, and offer a new way to detect and illuminate the effects of larger structural variants such as CNVs that impact the genome in complex ways.
“The potential to be able to look deeply at these variants gives us more opportunities to uncover ways in which genetic variation influences human phenotypes,” said Po-Ru Loh, senior author of the study, an associate member at the Broad, and an assistant professor at Brigham and Women’s Hospital and Harvard Medical School. “Downstream, it gives us more clues to be able to interpret and understand complex associations between genetics and trait variation.”
Capturing copy number variants
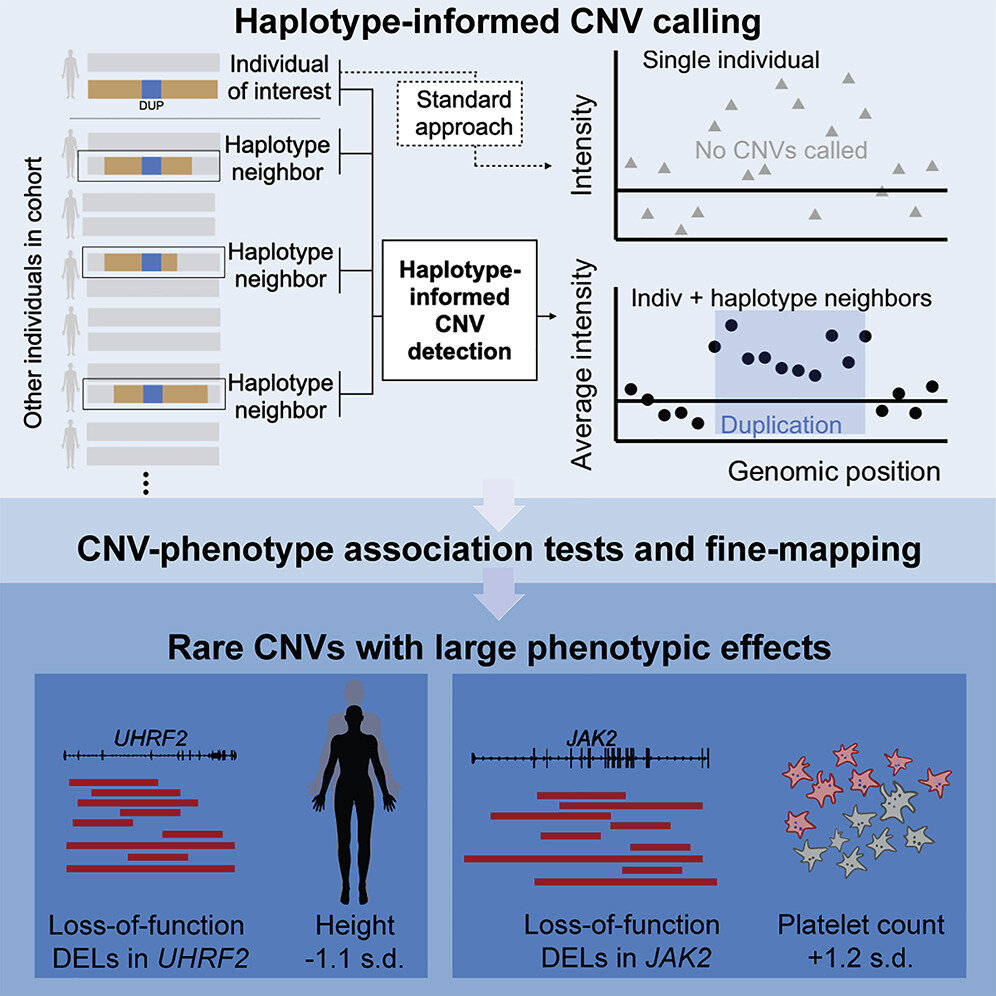
Many biobanks contain data on single nucleotide polymorphisms (SNPs), or single-letter changes in DNA, in large populations. Though very common, SNPs usually have at most slight effects on a trait. On the other hand, CNVs—which range from 50 to millions of base pairs in length—disable some genes and can induce more significant changes in the genome such as increasing the number of copies of a gene. Loh’s team wanted to improve detection of these structural changes from existing troves of SNP data, such as from the U.K. Biobank.
“In a lot of large cohorts, genetic variation has only been measured using SNP-array data, from which it is pretty hard to detect small CNVs using current algorithms. We thought that there might be other information in the cohorts that we could leverage to increase our ability to detect these CNVs,” said Margaux Hujoel, first author on the study and a postdoctoral researcher in Loh’s lab.
Hujoel and the team built an algorithm that grouped together the U.K. Biobank SNP probe intensity data of individuals who were distantly related to each other based on sharing a haplotype (a cluster of SNPs). This reduced noise in the data and enabled the detection of six times more CNVs than previous techniques. They found that the CNVs they detected accounted for half of all the gene inactivations scientists have previously attributed to structural changes in the genome.
The team then searched for associations between the CNVs and 56 traits. They identified more than 250 associations involving nearly 100 loci, or genomic regions, that were likely a direct result of CNVs. Many revealed new ties between specific genes and traits such as height. For example, individuals who had very rare CNVs that disabled the UHRF2 gene were, on average, about seven centimeters shorter than those who didn’t. Other rare variants with strong effects—discoverable only in large, biobank-scale cohorts—could offer valuable insights into genomic influences on complex disease.

Hidden secrets
Hujoel and Loh teamed up with Chikashi Terao, a group leader at the RIKEN Center for Integrative Medical Sciences who was a fellow postdoc at Broad and Brigham and Women’s Hospital with Loh, to apply their model to data from the BioBank Japan and confirmed many of the same trends. Loh hopes that other researchers will use their software to analyze genomic data in other biobanks. “This tool should be readily applicable for conducting the same sort of analysis in other ancestry groups, which could turn up quite different and interesting genetic associations,” he said.
The team says the large majority of CNVs are still left to be discovered, even in the U.K. Biobank. Because large biobanks have mostly generated SNP data using arrays that look at only certain locations in the genome, they miss most CNVs. Hujoel is in the process of adapting their method so that researchers can use it to study whole exome sequencing data, which examines all of the protein-coding regions of the genome. Loh also imagines that others might apply it to whole genome sequencing data to detect CNVs in the entire genome.
“There’s a lot of interest in exploring these more hidden parts of the genome that have been invisible to most genetic association studies to date,” Hujoel said. “We view our work as both a methodology that hopefully will continue to be useful and adaptable to other sources of data, and also as more motivation for people to continue delving into the ways that structural variation shapes human traits.”